Usage
One or more protein sequences can be input into the server in FASTA format. The server accepts at most 200 protein sequences per job. The user can either paste one or more sequences in the text-area provided, or, alternatively, upload a FASTA file containing protein sequences.
The BUSCA home page looks like the following:
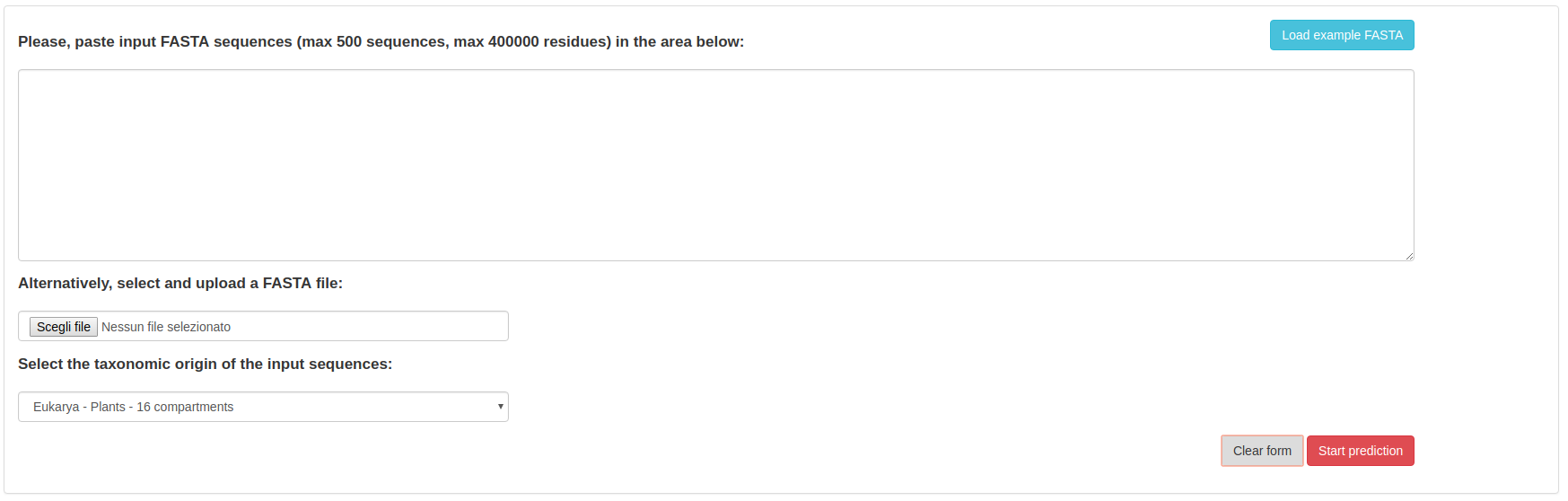
Besides the actual input sequences, the user must specify (through the designated selection menu) which organism class the sequences belong to, chosing among:
- Eukarya - Plants - for plant proteins
- Eukarya - Animals - for animal proteins
- Eukarya - Fungi - for fungi proteins
- Eukarya - Other - for any other eukaryotic protein which does not fit with any of the previuos
- Prokarya - Gram-negative - for prokaryotic proteins of gram-negative bacteria
- Prokarya - Gram-positive - for prokaryotic proteins of gram-positive bacteria or archaea
- Prokarya - Other - for any other prokaryotic protein
Example input sequences can be automatically loaded into the text-area using the "Example FASTA" button. To submit the job, the user should simply click on the "Start prediction" button.
Upon submission, the user will be automatically redirected to the page where the job results will be available after completion. If the job is still running, the result table shows no content.
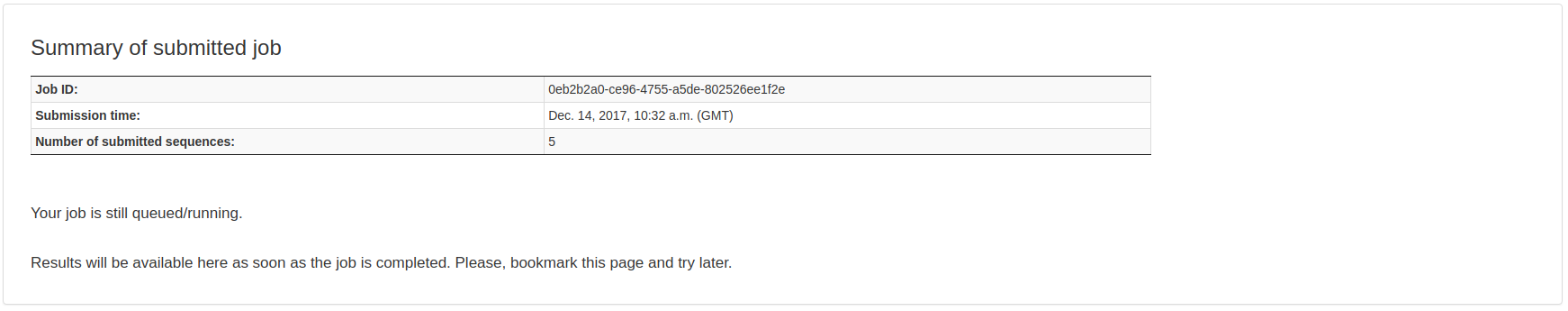
Output
The typical BUSCA output looks like the following:
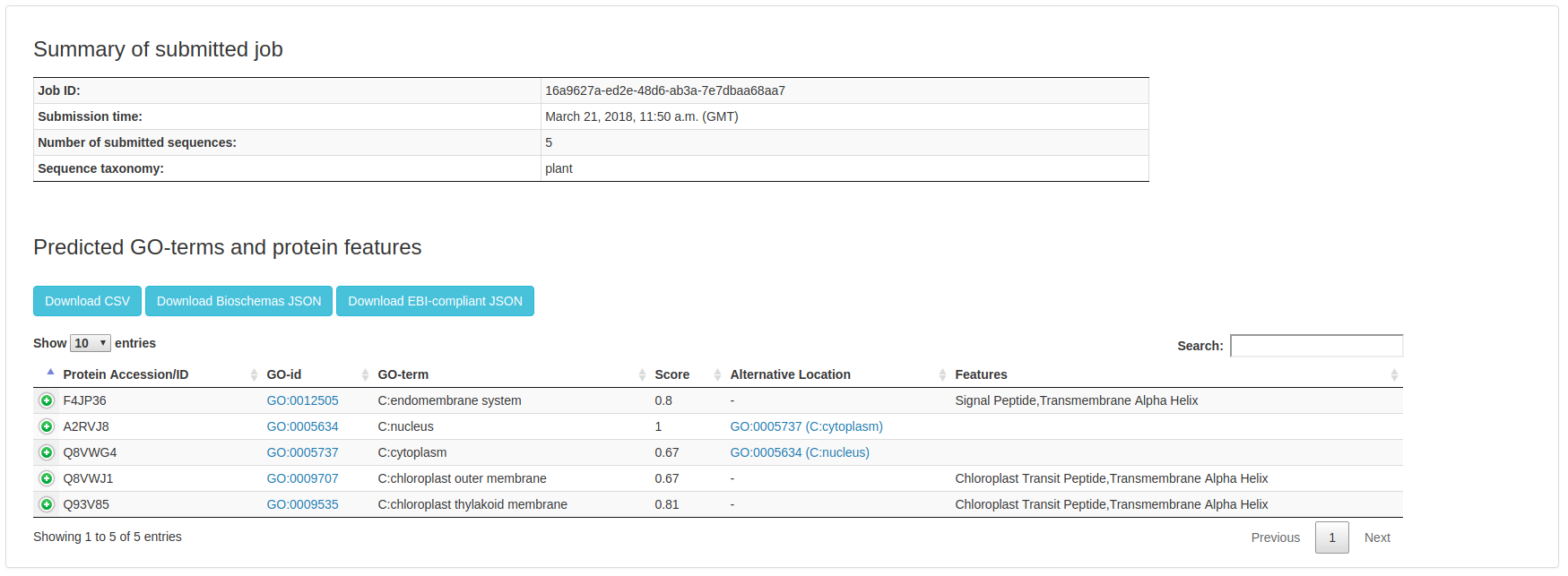
In the first section, main information about the job is reported.
Job information include:
- Job ID: a universal unique identifier which can be used to unambigously retrieve the job results after completion.
- Submission time: the GMT time of submission
- Number of submitted sequences: the total number of sequences included in the submission (either pasted in the text-area or uploaded as a file)
- Sequence taxonomy: the taxonomy the sequences belong to, as specified by the user at submission time
The section "Predicted GO-terms and protein featues" contains the actual prediction result, displayed as a paginated table. By default, 10 entries are shown on each page. Clicking on the pagination buttons, the user can browse results. For each submitted protein, the following data are reported:
- the protein identifier (as can be read from the input FASTA)
- the predicted GO-terms
- a prediction score associated to predicted GO-terms
- a potential alternative localization for the protein (if any)
- a summary of the features annotated along the sequence
By clicking on the expand details button (marked with the  icon and placed on the left of each protein accession), it is possible to visualize features predicted on that specific protein, if any. Different features are visualized on different annotation tracks along the sequence:
icon and placed on the left of each protein accession), it is possible to visualize features predicted on that specific protein, if any. Different features are visualized on different annotation tracks along the sequence:
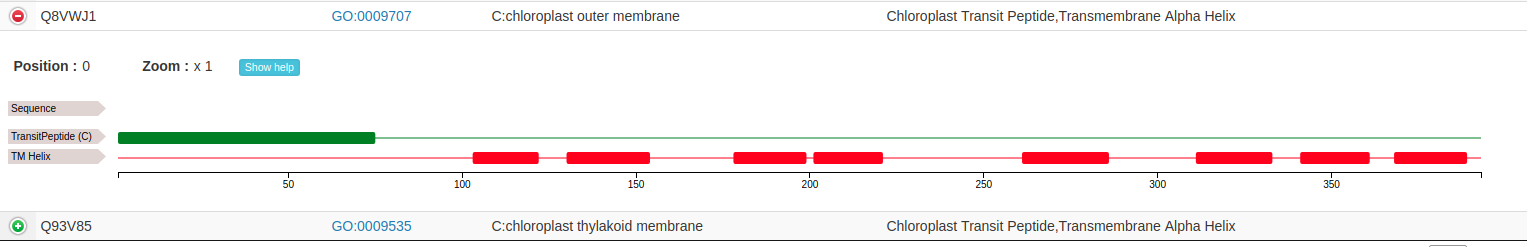
The user can zoom in by left-clicking and selecting the area of interest:
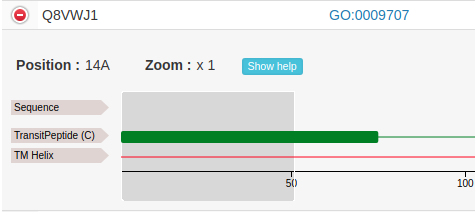
This allows to see feature annotation at residue-level:
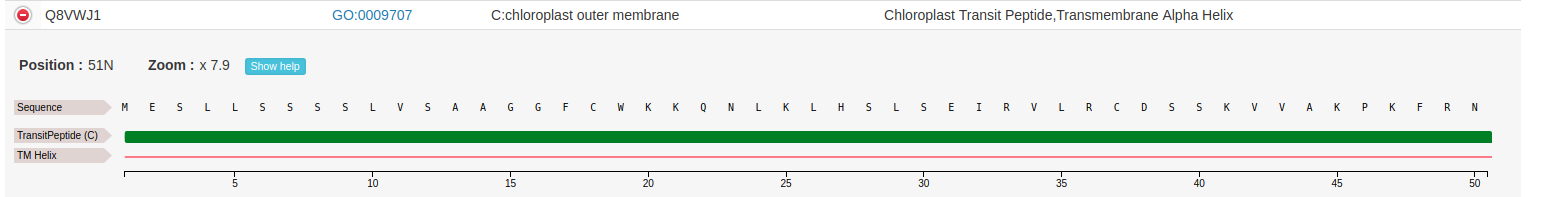
To zoom out just right-click on the feature track to reset the scale. Note that, sometimes, when the sequence is too long (as in the example shown), the actual residues are not visible at the initial default zoom level.
The feature panel can be closed by clicking on  . The button "Show help" provides an explaination on how to use the interactive feature panel.
. The button "Show help" provides an explaination on how to use the interactive feature panel.
The "Search" box can be used to free-text search the result table, displaying only entries meeting provided search criteria. Furthermore, result table can be sorted by any field by clicking on the corresponding field header.
Using the buttons "Download JSON" and "Download CSV" the user can download the job results in either JSON or CSV formats.